Općenitost
Osteogenesis imperfecta je urođena genetska bolest, koja nije povezana sa spolom, odgovorna je za određenu krhkost kostiju i izrazitu sklonost prijelomima.
Simptomi osteogeneze imperfecta su brojni; općenito se sastoje od: slabljenja kostiju, velike sklonosti prijelomima kostiju, prisutnosti plave, sive ili ljubičaste očne bjeloočnice, prisutnosti deformiteta kostiju ili drugih skeletnih promjena, trokutastog lica, krhkosti zuba itd. .
Općenito, sljedeće su bitne za ispravnu dijagnozu nedostatka osteogeneze: fizički pregled, povijest bolesti, medicinski slikovni testovi, test procjene kolagena tipa I i genetski test.
Nažalost, trenutno su jedini tretmani dostupni pacijentima s nedostatkom osteogeneze simptomatski. Dotična je bolest, naime, neizlječiva.
Što je osteogeneza imperfekta?
Osteogenesis imperfecta genetska je bolest koja kosti zahvaćene osobe čini slabijima i sklonijima prijelomima.
U stvarnosti, s pojmom osteogenesis imperfecta, liječnici upućuju na heterogenu skupinu genetskih bolesti, koju karakterizira određeni stupanj krhkosti kostiju. Stoga postoji nekoliko oblika (ili vrsta) osteogeneze imperfecta, neki mnogo teži od drugih.
TO JE UROĐENA BOLEST
U ljudi koji su zahvaćeni njome, osteogenesis imperfecta je bolest prisutna od rođenja, pa se prema svim namjerama može definirati kao urođena bolest.
JE LI Spol povezan?
Osteogenesis imperfecta nije rodno povezana genetska bolest, poput hemofilije ili Klinefelterovog sindroma.
EPIDEMILOGIJA
Prema nekim statističkim istraživanjima, učestalost osteogeneze imperfecta bila bi jednaka jednom slučaju na svakih 15.000-20.000 rođenih. To znači da svakih 15.000-20.000 novorođenčadi ima jedno oboljelo od nedostatka osteogeneze.
Druge statističke studije također su pokazale da osteogenesis imperfecta jednako pogađa muškarce i žene, te da nema sklonost prema određenoj populaciji ili etničkoj skupini.
Životni vijek iznimno je promjenjiv parametar, koji ovisi o obliku osteogeneze imperfecta.
Uzroci
Osteogeneza imperfecta gotovo uvijek proizlazi iz kvalitativne i kvantitativne promjene proizvodnje kolagena tipa I.
Kolagen tipa I neophodan je za jačanje kostiju i za održavanje zdravog vezivnog tkiva koje čini hrskavicu, tetive, kožu, očnu bjeloočnicu itd.
Stoga, promjena u proizvodnji kolagena tipa I utječe na čvrstoću kostiju i dobro zdravlje vezivnog tkiva prisutnog u ljudskom tijelu.
ŠTO MIJENJA PROIZVODNJU KOLAGENA?
Genetska bolest je stanje koje nastaje zbog mutacije jednog ili više gena koji čine staničnu DNK.
U slučaju osteogeneze imperfecta, uzroci potonjeg gotovo se uvijek mogu pronaći u mutaciji jednog ili oba gena COL1A1 (smješten na kromosomu 17) i COL1A2 (smješten na kromosomu 7).
U normalnim uvjetima, COL1A1 i COL1A2 reguliraju normalnu proizvodnju kolagena tipa I; u prisutnosti mutacija u svom naboju, one ne uspijevaju u regulatornoj funkciji.
Važno: koji drugi geni, ako su mutirani, uzrokuju osteogenezu imperfecta?
Osim mutacija COL1A1 i COL1A2, mutacije u genima IFITM5, SERPINF1, CRTAP i LEPRE1 potencijalni su uzroci osteogeneze imperfecta.
Spomenuti geni pokrivaju funkcije različite od COL1A1 i COL1A2 - stoga ne kontroliraju proizvodnju kolagena tipa I - ali i dalje imaju "utjecaj na čvrstoću i otpornost kostiju ljudskog kostura".
KAKVA JE GENETSKA BOLEST?
Osteogenesis imperfecta je autosomna genetska bolest.
Izraz autosomni, povezan s genetskom bolešću, ukazuje da je dotično stanje posljedica genetskih mutacija temeljenih na autosomnim i ne-spolnim kromosomima.
Podsjećamo čitatelje da ljudsko biće posjeduje kromosomski skup od 23 para ukupnih kromosoma, u kojima su 22 para autosomnog tipa, a samo jedan par spolnog tipa. Par kromosoma spolnog tipa utječe na spol pojedinac.
Nedostatak osteogeneze nakon mutacija u COL1A1, COL1A2 i IFITM5 ima sve karakteristike autosomno dominantne bolesti, a kada je posljedica mutacija u genima SERPINF1, CRTAP i LEPRE1, ima karakteristike autosomno recesivne bolesti.
VRSTE
Trenutačno liječnici vjeruju da postoji 8 vrsta (ili oblika) osteogeneze imperfecta. Kako bi razlikovali različite vrste, odlučili su se koristiti rimskim numeriranjem, točnije prvih osam rimskih brojeva.
Donja tablica prikazuje 8 oblika nedovršene osteogeneze, mutacije koje ih uzrokuju i druge karakteristike.
Momak
Mutirani gen
Vrsta genetske bolesti
THE
COL1A1
Autosomno dominantna
II
COL1A1 i COL1A2
Autosomno dominantna
III
COL1A1 i COL1A2
Autosomno dominantna
IV
COL1A1 i COL1A2
Autosomno dominantna
V.
IFITM5
Autosomno dominantna
VAS
SERPINF1
Autosomno recesivno
VII
CRTAP
Autosomno recesivno
VIII
HARE 1
Autosomno recesivno
* Napomena: očito su mutacije u COL1A1 i COL1A2, koje uzrokuju prva četiri oblika imperfekta osteogeneze, genetske promjene s nešto drugačijim karakteristikama. Inače ne bi imalo smisla razlikovati jedno od drugog.
Simptomi, znakovi i komplikacije
Sve vrste osteogeneze imperfecta odgovorne su za slabljenje kostiju, tako da osoba pogođena bolešću ima posebnu predispoziciju za prijelome. Stupanj slabljenja kostiju varira ovisno o obliku; za neke od njih ovo slabljenje je veće nego za druge.
Rekavši to, potrebno je istaknuti da svaki oblik osteogeneze imperfecta ima svoju simptomatsku sliku, koja se za neke može prisjetiti simptomatološke slike drugih oblika.
MOGUĆI SIMPTOMI I ZNACI
Mogući simptomi i znakovi nedostatne osteogeneze uključuju:
- Prisutnost malformacija kostiju;
- Prisutnost kratkog i malog tijela (namijenjeno kao prtljažnik);
- Problemi sa zglobovima (npr. Labavi zglobovi);
- Slabost mišića;
- Plava, ljubičasta ili siva očna bjeloočnica;
- Trokutasto lice;
- Bačvasti sanduk;
- Morfološke anomalije kralježaka;
- Zubna krhkost;
- Pad ili potpuni gubitak sluha;
- Problemi s disanjem
- Problemi povezani s nedostatkom ili nedostatkom kolagena tipa 1.
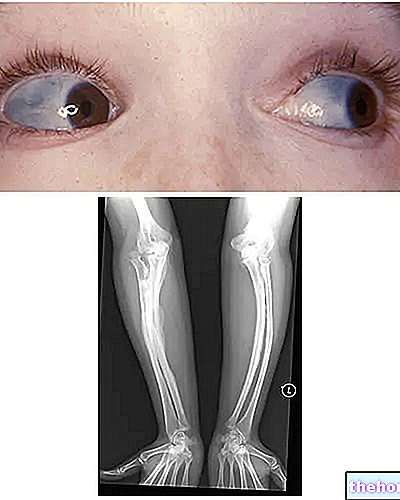
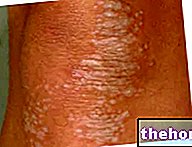
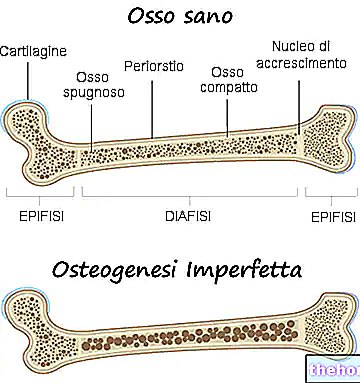
Osteogeneza nesavršena: obratite pozornost na plavo obojenje bjeloočnica i koštane deformacije koje karakteriziraju bolest. S wikipedia.org
KOJI SU NAJTEŽI OBLICI NEPRAVILNE OSTEOGENEZE?
Liječnici klasificiraju simptomatološku ozbiljnost različitih vrsta osteogeneze imperfecta na ljestvici od 3 stupnja, a to su: blagi stupanj, umjereni stupanj i teški stupanj.
Samo jedan oblik pripada kategoriji "blagi stupanj": "tip I osteogenesis imperfecta"; 4 oblika osteogenesis imperfecta spadaju u kategoriju "umjerenog stupnja": IV, V i VI; na kraju, u kategoriju "teškog stupnja" pripadaju 3 oblici: II, III, VII i VIII.
TIP I: OSOBINE
Najčešći i najteži oblik od svih, osteogenesis imperfecta tipa I ima sljedeće karakteristike:
- Izaziva prijelome osobito prije puberteta;
- Nema "gotovo nikakav utjecaj na visinu, pa su pacijenti obično normalne" visine;
- Uzrokuje probleme sa zglobovima i slabost mišića
- Odgovoran je za plavu, ljubičastu ili sivu bjeloočnicu;
- To je uzrok trokutastih anomalija lica i kralježnice;
- Gotovo nikada ne uzrokuje deformacije kostiju. Ako ih to izazove, minimalni su;
- Može uzrokovati krhkost zuba i / ili gubitak sluha (potonje se obično događa u odrasloj dobi);
- Povezan je s prisutnošću kolagena tipa I koji je normalne kvalitete, ali abnormalne količine (lošiji je od normalnog).
TIP II: OSOBINE
Osteogeneza imperfekta tipa II karakteriziraju:
- Uzrok smrti pri rođenju ili nedugo nakon toga. Problemi s disanjem gotovo uvijek uzrokuju smrt;
- Prisutnost značajne krhkosti kostiju i ozbiljnih koštanih deformacija;
- Pluća niskog rasta i nerazvijena
- Sklera plave, ljubičaste ili sive boje;
- Prisutnost kvantitativnih i kvalitativnih anomalija kolagena tipa I.
TIP III: OSOBINE
Osteogeneza nedovršenog tipa III ima sljedeće karakteristike:
- Iako vrlo ozbiljan, ne uzrokuje često smrt u razdoblju novorođenčadi;
- Povezan je s "visokom krhkošću kostiju;
- Odgovoran je za niski rast, probleme sa zglobovima, slabost mišića (osobito nogu i ruku), prsa u obliku cijevi, trokutasto lice i abnormalnu zakrivljenost kralježnice;
- To je zbog plave, ljubičaste ili sive bjeloočnice;
- Može uzrokovati probleme s disanjem, krhkost zubi i gubitak sluha;
- Često je odgovoran za deformacije kostiju;
- Povezan je s kvalitativnim i kvantitativnim abnormalnostima kolagena tipa I.
TIP IV: ZNAČAJKE
Osteogenezu tipa IV karakteriziraju:
- Stupanj krhkosti kosti između oblika II i III i oblika I;
- Niži rast od prosjeka;
- Sklera plave, ljubičaste ili sive boje;
- Deformiteti kostiju blage / umjerene cjeline, blage abnormalnosti kralježnice i prsnog koša;
- Trokutasto lice;
- Moguća prisutnost krhkosti zuba i gubitak sluha;
- Prisutnost abnormalnosti kolagena tipa I.
TIP V: ZNAČAJKE
Osteogeneza imperfekta tipa V na neki način nalikuje osteogenezi imperfekta tipa IV. Međutim, ima neke posebnosti, a to su:
- Sklera normalne boje;
- Odsutnost zubne krhkosti;
- Formiranje abnormalnih koštanih žuljeva tijekom procesa zarastanja slomljenih kostiju;
- Kalcifikacija međukoštane membrane koja se nalazi između radijusa i ulne. To narušava pokretljivost podlaktice.
TIP VI: OSOBINE
Također, osteogenesis imperfecta tipa VI slična je obliku IV. Razlikuju se od potonje neke posebnosti, uključujući visoku razinu alkalne fosfataze u krvi i prisutnost, na nekim kostima, lamela (koštanih) sličnih bodlji ribe.
TIP VII: OSOBINE
Simptomatski, osteogenesis imperfecta tipa VII može u nekim okolnostima nalikovati na tip IV, a u drugim na tip II.
Osobitosti ovog ozbiljnog patološkog oblika uključuju:
- Nizak rast;
- Prisutnost izuzetno kratkog humerusa (kosti ruke) i bedrene kosti (kosti bedra);
- Česta prisutnost deformiteta kuka, poznatog kao coxa vara.
TIP VIII: KARAKTERISTIKE
Osteogeneza imperfekta VIII. Tipa jako podsjeća na oblike II i III.
Među njegovim posebnim karakteristikama ističu se sljedeće: ozbiljan deficit rasta, teška skeletna hipomineralizacija i odsutnost (ili oskudna prisutnost) enzima prolil 3-hidroksilaze.
Dijagnoza
Općenito, dijagnostički proces kojem su podvrgnuti pacijenti kod kojih postoji sumnja na oblik osteogeneze imperfecta započinje pažljivim fizičkim pregledom i pomnom anamnezom; zatim se nastavlja "analizom obiteljske povijesti pacijenta i s nizom dijagnostičkih slikovnih testova (X-zrake, CT snimke itd.); konačno, završava kvantitativnom i kvalitativnom procjenom kolagena tipa I i genetski test.
Danas postoji mogućnost dijagnosticiranja osteogeneze imperfecta čak i u prenatalnoj fazi, podvrgavanjem trudnice ultrazvuku.
ZNAČAJ OBJEKTIVNOG ISPITIVANJA I POVIJEST
Liječnik stručnjak za osteogenezu imperfecta vrlo je često u stanju dijagnosticirati spomenutu bolest čak i fizičkim pregledom i anamnezom. To znači da ovi dijagnostički testovi nemaju zanemarivu važnost.
OCJENA PROIZVODNJE KOLAGENA TIPA I
U pravilu je kvalitativna i kvantitativna ocjena kolagena tipa I vrlo pouzdan test, jer je, kako je navedeno, većinu slučajeva imperfekta osteogeneze karakteriziraju mutacije u genima koji kontroliraju proizvodnju kolagena tipa 1.
Kako bi procijenili količinu i kvalitetu kolagena tipa I prisutnog na staničnoj razini u pojedinca, liječnici se mogu osloniti na biopsiju kože ili određeni krvni test.
Oba ova evaluacijska testa prilično su složena i pacijent (ili njegovi roditelji) će možda morati pričekati nekoliko tjedana da sazna rezultate.
GENETIČKI TEST
Genetskim testom koji ispituje cijelu DNK ispitane osobe liječnici mogu definitivno odrediti karakteristike prisutne genetske mutacije.
Općenito, izvođenje genetskog testa na cijeloj staničnoj DNK predviđeno je kada procjena karakteristika kolagena tipa I nije dala željene rezultate ili ako nije riječ o mutaciji u COL1A1 ili COL1A2 koja uzrokuje „osteogenezu imperfecta.
PRENATALNA DIJAGNOSTIKA
Prenatalni ultrazvuk vrlo je koristan za identifikaciju nedostatka osteogeneze tipa II i III.
Terapija
Trenutačno ne postoji poseban lijek za osteogenezu imperfecta. Drugim riječima, osobama s nedostatkom osteogeneze suđeno je živjeti s navedenim stanjem do smrti, što je često posljedica same bolesti.
Nedostatak specifične terapije ne isključuje postojanje drugih oblika liječenja. Zapravo, među terapijskim mogućnostima bolesnika s imperfektom osteogeneze uključene su različite simptomatske terapije; pod simptomatskim terapijama mislimo na tretmane sposobne ublažiti simptome, usporiti tijek bolesti i spriječiti (ili barem odgoditi) najteže posljedice.
MOGUĆI SIMPTOMATSKI TRETMANI
Na popisu mogućih simptomatskih tretmana za osteogenezu imperfecta ističu se:
- Kirurško umetanje noktiju, unutar najdužih kostiju (N.B .: najsklonije prijelomima), pruža veću otpornost na prijelome i deformitete. Ova operacija se naziva rodding intramedularno;
- Konzervativno ili kirurško liječenje prijeloma i / ili deformacija kostiju;
- Zubna njega, za zaštitu zdravlja zubi;
- Terapije za ublažavanje boli, u slučaju vrlo bolnih višestrukih prijeloma;
- Fizioterapija, za produljenje i jačanje mišića.Elastični i tonički mišićni aparat omogućuje vam sprječavanje padova koji bi mogli dovesti do različitih prijeloma kostiju;
- Upotreba pomagala za kretanje, uključujući invalidska kolica, proteze, štake itd.
KORISTI KRETANJA
Za osobe s nedostatkom osteogeneze, liječnici preporučuju stalnu praksu tjelesnih vježbi i kretanja općenito, jer obje ove aktivnosti doprinose jačanju koštanog i mišićnog sustava.
Među preporučenim sportovima su: plivanje, budući da je to "tjelesna aktivnost niskog utjecaja na koštani sustav", te hodanje.
KORISTI ZDRAVOG NAČINA ŽIVOTA
Zdrav život, izbjegavanje pušenja, pijenje viška alkohola, prekomjerno i loše jelo itd. Ima više nego diskretne zdravstvene koristi za pacijente s nedostatkom osteogeneze, jer usporava napredovanje bolesti i smanjuje krhkost kostiju.
SIMPTOMATSKI TRETMANI U FAZI EKSPERIMENTACIJE
Trenutno liječnici i istraživači procjenjuju učinkovitost nekih simptomatskih tretmana, uključujući liječenje hormonima rasta i intravenoznu i oralnu terapiju na bazi bisfosfonata.
Za sada, rezultati gore spomenutih istražnih tretmana služe dobro cijeloj medicinskoj zajednici.
Prognoza
Osteogenesis imperfecta je bolest s negativnom prognozom, jer je neizlječiva, drastično ugrožava kvalitetu života i, u nekim slučajevima, uzrokuje preranu smrt oboljelog subjekta.
Međutim, treba napomenuti da su, također zahvaljujući modernim simptomatskim tretmanima, mnoge osobe s blagim oblikom osteogeneze imperfecta sposobne voditi ugodan i zadovoljavajući život.
Prevencija
Nažalost, trenutno nema preventivnih mjera protiv osteogeneze imperfecta.